Entwicklung des Embryos. Epidemiologische und demografische Daten zur menschlichen Embryonalentwicklung
Die Untersuchung der Entwicklung des menschlichen Körpers von der Bildung einer einzelligen Zygote oder befruchteten Eizelle bis zur Geburt eines Kindes. Die embryonale (intrauterine) menschliche Entwicklung dauert etwa 265–270 Tage. In dieser Zeit bilden sich aus der ursprünglichen Zelle mehr als 200 Millionen Zellen und die Größe des Embryos wächst von mikroskopisch auf einen halben Meter.
Generell lässt sich die Entwicklung eines menschlichen Embryos in drei Phasen einteilen. Der erste ist der Zeitraum von der Befruchtung der Eizelle bis zum Ende der zweiten Woche des intrauterinen Lebens, wenn sich der sich entwickelnde Embryo (Embryo) in die Gebärmutterwand einnistet und beginnt, Nahrung von der Mutter zu erhalten. Die zweite Etappe dauert von der dritten bis zum Ende der achten Woche. In dieser Zeit werden alle wichtigen Organe gebildet und der Embryo erhält die Merkmale eines menschlichen Körpers. Am Ende der zweiten Entwicklungsstufe spricht man bereits von einer Frucht. Die Länge des dritten Stadiums, manchmal auch fetal genannt (vom lateinischen fetus – Fötus), reicht vom dritten Monat bis zur Geburt. In diesem letzten Stadium ist die Spezialisierung der Organsysteme abgeschlossen und der Fötus erlangt nach und nach die Fähigkeit, unabhängig zu existieren.
Genitzellen und Befruchtung
Beim Menschen ist eine reife Fortpflanzungszelle (Gamete) beim Mann ein Spermium, bei der Frau eine Eizelle. Bevor Gameten zu einer Zygote verschmelzen, müssen sich diese Geschlechtszellen bilden, reifen und sich dann treffen.
Menschliche Keimzellen ähneln in ihrer Struktur den Gameten der meisten Tiere. Der grundlegende Unterschied zwischen Gameten und anderen Körperzellen, sogenannten somatischen Zellen, besteht darin, dass ein Gamet nur die Hälfte der Chromosomen einer somatischen Zelle enthält. In menschlichen Keimzellen gibt es 23 davon. Während des Befruchtungsprozesses bringt jede Keimzelle ihre 23 Chromosomen in die Zygote ein, und somit verfügt die Zygote über 46 Chromosomen, also einen doppelten Satz davon, wie es allen menschlichen Körpern innewohnt Zellen. Siehe auch ZELLE.
Obwohl Spermien und Eizellen in ihren wesentlichen Strukturmerkmalen somatischen Zellen ähneln, sind sie gleichzeitig hochspezialisiert auf ihre Rolle bei der Fortpflanzung. Ein Spermium ist eine kleine und sehr bewegliche Zelle (siehe SPERMA). Die Eizelle hingegen ist unbeweglich und viel größer (fast 100.000 Mal) als das Sperma. Der größte Teil seines Volumens besteht aus Zytoplasma, das Nährstoffreserven enthält, die der Embryo während der ersten Entwicklungsphase benötigt (siehe EGG).
Für die Befruchtung müssen Eizelle und Spermium reif werden. Darüber hinaus muss die Eizelle innerhalb von 12 Stunden nach Verlassen des Eierstocks befruchtet werden, sonst stirbt sie ab. Menschliche Spermien leben länger, etwa einen Tag. Mit Hilfe seines peitschenförmigen Schwanzes gelangt das Sperma schnell in den mit der Gebärmutter verbundenen Gang – den Eileiter, in den die Eizelle vom Eierstock gelangt. Dies dauert normalerweise weniger als eine Stunde nach der Kopulation. Es wird angenommen, dass die Befruchtung im oberen Drittel des Eileiters stattfindet.
Obwohl das Ejakulat normalerweise Millionen von Spermien enthält, dringt nur eines in die Eizelle ein und löst eine Kette von Prozessen aus, die zur Entwicklung des Embryos führen. Aufgrund der Tatsache, dass das gesamte Sperma in die Eizelle eindringt, bringt der Mann dem Nachwuchs neben Kernmaterial auch eine bestimmte Menge zytoplasmatischen Materials mit, darunter das Zentrosom, eine kleine Struktur, die für die Zellteilung der Zygote notwendig ist. Das Sperma bestimmt auch das Geschlecht des Nachwuchses. Als Höhepunkt der Befruchtung gilt der Zeitpunkt der Verschmelzung des Spermienkerns mit dem Eizellenkern.
ZERSTÖRUNG UND IMPLANTATION
Nach der Befruchtung wandert die Zygote allmählich durch den Eileiter in die Gebärmutterhöhle. Während dieses Zeitraums, über einen Zeitraum von etwa drei Tagen, durchläuft die Zygote eine Phase der Zellteilung, die als Spaltung bezeichnet wird. Bei der Fragmentierung nimmt die Anzahl der Zellen zu, ihr Gesamtvolumen ändert sich jedoch nicht, da jede Tochterzelle kleiner ist als die ursprüngliche. Die erste Spaltung erfolgt etwa 30 Stunden nach der Befruchtung und führt zu zwei völlig identischen Tochterzellen. Die zweite Spaltung erfolgt 10 Stunden nach der ersten und führt zur Bildung eines Vierzellstadiums. Etwa 50–60 Stunden nach der Befruchtung ist das sogenannte Stadium erreicht. Morula – eine Kugel aus 16 oder mehr Zellen.
Mit fortschreitender Spaltung teilen sich die äußeren Zellen der Morula schneller als die inneren Zellen, was dazu führt, dass die äußere Zellschicht (Trophoblast) vom inneren Zellhaufen (der sogenannten inneren Zellmasse) getrennt wird und nur noch mit ihnen in Verbindung bleibt ein Platz. Zwischen den Schichten bildet sich ein Hohlraum, das Blastocoel, der sich nach und nach mit Flüssigkeit füllt. In diesem Stadium, das drei bis vier Tage nach der Befruchtung auftritt, endet die Spaltung und der Embryo wird Blastozyste oder Blastula genannt. In den ersten Tagen der Entwicklung erhält der Embryo Nahrung und Sauerstoff aus den Sekreten des Eileiters.
Ungefähr fünf bis sechs Tage nach der Befruchtung, wenn sich die Blastula bereits in der Gebärmutter befindet, bildet der Trophoblast fingerartige Zotten, die unter kräftiger Bewegung beginnen, in das Uterusgewebe einzudringen. Gleichzeitig regt die Blastula offenbar die Produktion von Enzymen an, die die teilweise Verdauung der Gebärmutterschleimhaut (Endometrium) fördern. Etwa am 9.–10. Tag implantiert (wächst) der Embryo in die Gebärmutterwand und ist vollständig von seinen Zellen umgeben; Mit der Einnistung des Embryos stoppt der Menstruationszyklus.
Zusätzlich zu seiner Rolle bei der Einnistung ist der Trophoblast auch an der Bildung des Chorions beteiligt, der primären Membran, die den Embryo umgibt. Das Chorion trägt wiederum zur Bildung der Plazenta bei, einer Membran mit schwammiger Struktur, über die der Embryo anschließend Nahrung erhält und Stoffwechselprodukte abtransportiert.
Embryonale Keimschichten
Der Embryo entwickelt sich aus der inneren Zellmasse der Blastula. Wenn der Flüssigkeitsdruck im Blastocoel zunimmt, verdichten sich die Zellen der inneren Zellmasse und bilden den Keimschild oder das Blastoderm. Der Embryonalschild ist in zwei Schichten unterteilt. Einer von ihnen wird zur Quelle der drei primären Keimschichten: Ektoderm, Endoderm und Mesoderm. Der Prozess der Trennung der ersten beiden und dann der dritten Keimschicht (die sogenannte Gastrulation) markiert die Umwandlung der Blastula in die Gastrula.
Die Keimschichten unterscheiden sich zunächst nur in der Lage: Das Ektoderm ist die äußerste Schicht, das Endoderm die innere Schicht und das Mesoderm die Zwischenschicht. Die Bildung der drei Keimblätter ist etwa eine Woche nach der Befruchtung abgeschlossen.
Nach und nach, Schritt für Schritt, entstehen aus jeder Keimschicht bestimmte Gewebe und Organe. Somit bildet das Ektoderm die äußere Schicht der Haut und ihrer Derivate (Anhangsgebilde) – Haare, Nägel, Hautdrüsen, Mund-, Nasen- und Afterschleimhaut – sowie das gesamte Nervensystem und Sinnesorganrezeptoren, wie beispielsweise die Netzhaut . Aus dem Endoderm werden gebildet: Lungen; die Auskleidung (Schleimhaut) des gesamten Verdauungstrakts mit Ausnahme von Mund und Anus; einige an diesen Trakt angrenzende Organe und Drüsen wie Leber, Bauchspeicheldrüse, Thymusdrüse, Schilddrüse und Nebenschilddrüse; Auskleidung der Blase und der Harnröhre. Mesoderm ist die Quelle des Kreislaufsystems, des Ausscheidungs-, Fortpflanzungs-, hämatopoetischen und Immunsystems sowie des Muskelgewebes, aller Arten von unterstützendem trophischem Gewebe (Skelett-, Knorpel-, lockeres Bindegewebe usw.) und der inneren Hautschichten ( Dermis). Ausgewachsene Organe bestehen meist aus mehreren Gewebetypen und sind daher aufgrund ihrer Herkunft unterschiedlichen Keimschichten zugeordnet. Aus diesem Grund ist es nur möglich, die Beteiligung der einen oder anderen Keimschicht am Prozess der Gewebebildung zu verfolgen.
EXTRAGEMONY-MEMBRANEN
Die Entwicklung des Embryos geht mit der Bildung mehrerer Membranen einher, die ihn umgeben und bei der Geburt abgestoßen werden. Das äußerste davon ist das bereits erwähnte Chorion, ein Derivat des Trophoblasten. Es ist durch einen aus dem Mesoderm stammenden Körperstiel aus Bindegewebe mit dem Embryo verbunden. Mit der Zeit verlängert sich der Stiel und bildet die Nabelschnur (Nabelschnur), die den Embryo mit der Plazenta verbindet.
Die Plazenta entwickelt sich als spezialisierter Auswuchs der Membranen. Die Chorionzotten durchstoßen das Endothel der Blutgefäße der Gebärmutterschleimhaut und tauchen in die mit mütterlichem Blut gefüllten Blutlücken ein. Somit ist das Blut des Fötus vom Blut der Mutter nur durch die dünne äußere Membran des Chorions und die Wände der Kapillaren des Embryos selbst getrennt, d. h. eine direkte Vermischung des Blutes der Mutter und des Fötus ist nicht möglich geschehen. Nährstoffe, Sauerstoff und Stoffwechselprodukte diffundieren durch die Plazenta. Bei der Geburt wird die Plazenta als Nachgeburt verworfen und ihre Funktionen werden auf das Verdauungssystem, die Lunge und die Nieren übertragen.
Im Chorion befindet sich der Embryo in einem Beutel namens Amnion, der aus dem embryonalen Ektoderm und Mesoderm gebildet wird. Die Fruchtblase ist mit Flüssigkeit gefüllt, die den Embryo mit Feuchtigkeit versorgt, ihn vor Stößen schützt und ihn in einem Zustand nahe der Schwerelosigkeit hält.
Eine weitere zusätzliche Schale ist die Allantois, eine Ableitung von Endoderm und Mesoderm. Dies ist der Lagerort für Ausscheidungsprodukte; Es verbindet sich mit dem Chorion im Körperstiel und fördert die Atmung des Embryos.
Der Embryo hat eine weitere temporäre Struktur – die sogenannte. Dottersack. Im Laufe der Zeit versorgt der Dottersack den Embryo durch Diffusion aus dem mütterlichen Gewebe mit Nährstoffen; Später werden hier Vorläufer-(Stamm-)Blutzellen gebildet. Der Dottersack ist der primäre Ort der Hämatopoese im Embryo; anschließend geht diese Funktion zunächst auf die Leber und dann auf das Knochenmark über.
EMBRYO-ENTWICKLUNG
Während der Bildung extraembryonaler Membranen entwickeln sich die Organe und Systeme des Embryos weiter. Zu bestimmten Zeitpunkten beginnt sich ein Teil der Zellen der Keimblätter schneller zu teilen als der andere, Zellgruppen wandern und Zellschichten ändern ihre räumliche Konfiguration und Position im Embryo. Zu bestimmten Zeiten ist das Wachstum einiger Zelltypen sehr aktiv und sie nehmen an Größe zu, während andere langsam wachsen oder ganz aufhören zu wachsen.
Das Nervensystem ist das erste, das sich nach der Implantation entwickelt. Während der zweiten Entwicklungswoche nimmt die Zahl der ektodermalen Zellen auf der Rückseite des Keimschildes rasch zu, wodurch sich über dem Schild eine Ausbuchtung bildet – der Primitivstreifen. Dann entsteht darauf eine Rille, in deren Vorderseite eine kleine Grube entsteht. Vor dieser Fossa teilen sich die Zellen schnell und bilden den Kopffortsatz, den Vorläufer des sogenannten. Rückensaite oder Akkord. Durch die Verlängerung bildet die Chorda dorsalis im Embryo eine Achse, die die Grundlage für die symmetrische Struktur des menschlichen Körpers bildet. Über der Chorda dorsalis befindet sich die Neuralplatte, aus der das Zentralnervensystem gebildet wird. Etwa am 18. Tag beginnt das Mesoderm an den Rändern der Chorda dorsalis, dorsale Segmente (Somiten) zu bilden, paarige Gebilde, aus denen sich die tiefen Schichten der Haut, der Skelettmuskulatur und der Wirbel entwickeln.
Nach dreiwöchiger Entwicklung beträgt die durchschnittliche Länge des Embryos vom Scheitel bis zum Schwanz nur noch etwas mehr als 2 mm. Dennoch sind bereits die Rudimente der Chorda und des Nervensystems sowie Augen und Ohren vorhanden. Es gibt bereits ein S-förmiges Herz, das pulsiert und Blut pumpt.
Nach der vierten Woche beträgt die Länge des Embryos etwa 5 mm, der Körper ist C-förmig. Das Herz, das die größte Ausbuchtung an der Innenseite der Körperwölbung bildet, beginnt sich in Kammern zu unterteilen. Es werden drei Hauptbereiche des Gehirns (Hirnbläschen) sowie die Seh-, Hör- und Riechnerven gebildet. Das Verdauungssystem wird gebildet, einschließlich Magen, Leber, Bauchspeicheldrüse und Darm. Die Strukturierung des Rückenmarks beginnt und es sind kleine paarige Gliedmaßenrudimente zu erkennen.
Ein vierwöchiger menschlicher Embryo weist bereits Kiemenbögen auf, die den Kiemenbögen eines Fischembryos ähneln. Sie verschwinden bald, aber ihr vorübergehendes Auftreten ist ein Beispiel für die Ähnlichkeit der Struktur des menschlichen Embryos mit anderen Organismen (siehe auch EMBRYOLOGIE).
Im Alter von fünf Wochen hat der Embryo einen Schwanz und die sich entwickelnden Arme und Beine ähneln Stümpfen. Muskeln und Verknöcherungszentren beginnen sich zu entwickeln. Der Kopf ist der größte Teil: Das Gehirn wird bereits durch fünf Hirnbläschen (Hohlräume mit Flüssigkeit) repräsentiert; Es gibt auch hervortretende Augen mit Linsen und pigmentierter Netzhaut.
Im Zeitraum von der fünften bis achten Woche endet die eigentliche Embryonalperiode der intrauterinen Entwicklung. In dieser Zeit wächst der Embryo von 5 mm auf etwa 30 mm und beginnt, einem Menschen zu ähneln. Sein Aussehen verändert sich wie folgt: 1) Die Krümmung des Rückens nimmt ab, der Schwanz wird weniger auffällig, teils aufgrund der Verkleinerung, teils weil er vom sich entwickelnden Gesäß verdeckt wird; 2) Der Kopf richtet sich auf, die äußeren Teile der Augen, Ohren und Nase erscheinen auf dem sich entwickelnden Gesicht; 3) Die Arme unterscheiden sich von den Beinen, man sieht bereits die Finger und Zehen; 4) die Nabelschnur ist vollständig definiert, der Bereich ihrer Befestigung am Bauch des Embryos wird kleiner; 5) Im Bauchbereich wächst die Leber stark und wird so konvex wie das Herz, und beide Organe bilden bis zur achten Woche ein klumpiges Profil des mittleren Körperteils; gleichzeitig macht sich der Darm in der Bauchhöhle bemerkbar, wodurch der Magen runder wird; 6) Der Hals wird vor allem dadurch, dass sich das Herz nach unten bewegt, sowie durch das Verschwinden der Kiemenbögen besser erkennbar. 7) Äußere Genitalien erscheinen, obwohl sie ihr endgültiges Aussehen noch nicht vollständig erreicht haben.
Bis zum Ende der achten Woche sind fast alle inneren Organe gut ausgebildet und die Nerven und Muskeln sind so weit entwickelt, dass der Embryo spontane Bewegungen ausführen kann. Von diesem Zeitpunkt an bis zur Geburt hängen die wichtigsten Veränderungen des Fötus mit Wachstum und weiterer Spezialisierung zusammen.
ABSCHLUSS DER FÖTALEN ENTWICKLUNG
In den letzten sieben Monaten der Entwicklung nimmt das Gewicht des Fötus von 1 g auf etwa 3,5 kg zu und die Länge nimmt von 30 mm auf etwa 51 cm zu. Die Größe des Babys zum Zeitpunkt der Geburt kann je nach Vererbung erheblich variieren , Ernährung und Gesundheit.
Während der Entwicklung des Fötus verändern sich nicht nur seine Größe und sein Gewicht, sondern auch die Körperproportionen stark. Bei einem zwei Monate alten Fötus beispielsweise ist der Kopf fast halb so lang wie der Körper. In den verbleibenden Monaten wächst es weiter, allerdings langsamer, sodass es bei der Geburt nur noch ein Viertel der Körperlänge ausmacht. Der Hals und die Gliedmaßen werden länger, während die Beine schneller wachsen als die Arme. Andere äußere Veränderungen stehen im Zusammenhang mit der Entwicklung der äußeren Genitalien, dem Wachstum von Körperhaaren und Nägeln; Durch die Ablagerung von Unterhautfett wird die Haut glatter.
Eine der bedeutendsten inneren Veränderungen ist mit dem Ersatz von Knorpel durch Knochenzellen während der Bildung eines reifen Skeletts verbunden. Die Fortsätze vieler Nervenzellen sind mit Myelin (einem Protein-Lipid-Komplex) bedeckt. Der Prozess der Myelinisierung führt zusammen mit der Bildung von Verbindungen zwischen Nerven und Muskeln zu einer erhöhten Beweglichkeit des Fötus in der Gebärmutter. Diese Bewegungen sind für die Mutter etwa nach dem vierten Monat deutlich zu spüren. Nach dem sechsten Monat dreht sich der Fötus in der Gebärmutter, sodass sein Kopf nach unten zeigt und auf dem Gebärmutterhals ruht.
Im siebten Monat ist der Fötus vollständig mit Vernix bedeckt, einer weißlichen Fettmasse, die nach der Geburt verschwindet. Für ein zu früh geborenes Kind ist es in dieser Zeit schwieriger zu überleben. In der Regel gilt: Je normaler die Geburt verläuft, desto größer sind die Überlebenschancen des Kindes, da der Fötus in den letzten Wochen der Schwangerschaft durch Antikörper aus dem Blut der Mutter vorübergehend vor bestimmten Krankheiten geschützt wird. Obwohl die Geburt das Ende der intrauterinen Periode markiert, setzt sich die biologische Entwicklung des Menschen im Kindes- und Jugendalter fort.
SCHÄDIGE AUSWIRKUNGEN AUF DEN FET
Geburtsfehler können verschiedene Ursachen haben, etwa Krankheiten, genetische Anomalien und zahlreiche schädliche Substanzen, die sich auf den Fötus und die Mutter auswirken. Kinder mit Geburtsfehlern können aufgrund körperlicher oder geistiger Behinderung lebenslang behindert sein. Das wachsende Wissen über die Verletzlichkeit des Fötus, insbesondere in den ersten drei Monaten, in denen sich seine Organe bilden, hat nun zu einer verstärkten Aufmerksamkeit für die vorgeburtliche Zeit geführt.
Krankheiten. Eine der häufigsten Ursachen für Geburtsfehler ist die Viruserkrankung Röteln. Erkrankt eine Mutter in den ersten drei Monaten der Schwangerschaft an Röteln, kann dies zu irreparablen Störungen in der Entwicklung des Fötus führen. Kleinkindern wird manchmal die Rötelnimpfung verabreicht, um das Risiko zu verringern, dass schwangere Frauen, die mit ihnen in Kontakt kommen, an der Krankheit erkranken. Siehe auch RUBELLA.
Auch sexuell übertragbare Krankheiten sind potenziell gefährlich. Syphilis kann von der Mutter auf den Fötus übertragen werden, was zu Fehlgeburten und Totgeburten führen kann. Eine festgestellte Syphilis muss umgehend mit Antibiotika behandelt werden, was für die Gesundheit der Mutter und ihres ungeborenen Kindes wichtig ist.
Eine fetale Erythroblastose kann beim Neugeborenen zu Totgeburten oder schwerer Anämie mit der Entwicklung einer geistigen Behinderung führen. Die Krankheit tritt bei Rh-Inkompatibilität zwischen dem Blut der Mutter und des Fötus auf (normalerweise bei einer wiederholten Schwangerschaft mit einem Rh-positiven Fötus). Siehe auch BLUT.
Eine weitere Erbkrankheit ist die Mukoviszidose, deren Ursache eine genetisch bedingte Stoffwechselstörung ist, die vor allem die Funktion aller exokrinen Drüsen (Schleim-, Schweiß-, Speicheldrüse, Bauchspeicheldrüse und andere) beeinträchtigt: Sie beginnen, extrem zähen Schleim zu produzieren, der beide verstopfen kann die Gänge selbst, Drüsen, die sie daran hindern, Sekrete abzusondern, und kleine Bronchien; Letzteres führt zu einer schweren Schädigung des bronchopulmonalen Systems und schließlich zur Entwicklung eines Atemversagens. Bei manchen Patienten ist vor allem die Aktivität des Verdauungssystems gestört. Die Krankheit wird kurz nach der Geburt entdeckt und führt beim Neugeborenen manchmal bereits am ersten Lebenstag zu einem Darmverschluss. Einige Manifestationen dieser Krankheit sind einer medikamentösen Therapie zugänglich. Galaktosämie ist ebenfalls eine Erbkrankheit, die durch das Fehlen eines Enzyms verursacht wird, das für den Stoffwechsel von Galaktose (einem Produkt der Verdauung von Milchzucker) notwendig ist, und zur Bildung von Katarakten sowie zu Schäden an Gehirn und Leber führt. Galaktosämie war bis vor Kurzem eine häufige Ursache für die Kindersterblichkeit, inzwischen wurden jedoch Methoden zur frühzeitigen Diagnose und Behandlung durch eine spezielle Diät entwickelt. Das Down-Syndrom (siehe DOWN-SYNDROM) wird in der Regel durch das Vorhandensein eines zusätzlichen Chromosoms in Zellen verursacht. Eine Person mit dieser Erkrankung ist normalerweise kleinwüchsig, hat leicht schräge Augen und eingeschränkte geistige Fähigkeiten. Die Wahrscheinlichkeit, dass ein Kind ein Down-Syndrom hat, steigt mit dem Alter der Mutter. Phenylketonurie ist eine Krankheit, die durch das Fehlen eines Enzyms verursacht wird, das für den Stoffwechsel einer bestimmten Aminosäure erforderlich ist. Es kann auch eine Ursache für geistige Behinderung sein (siehe PHENYLKETONURIE).
Einige Geburtsfehler können durch eine Operation teilweise oder vollständig korrigiert werden. Dazu gehören Muttermale, Klumpfüße, Herzfehler, zusätzliche oder verwachsene Finger und Zehen, Anomalien in der Struktur der äußeren Genitalien und des Urogenitalsystems, Spina bifida, Lippen- und Gaumenspalten. Zu den Defekten zählen auch die Pylorusstenose, d. h. eine Verengung des Übergangs vom Magen zum Dünndarm, das Fehlen von Anus und Hydrozephalus – ein Zustand, bei dem sich überschüssige Flüssigkeit im Schädel ansammelt, was zu einer Vergrößerung und Verformung des Kopfes führt geistige Behinderung (siehe auch ANGENITALE LUSTER).
Medikamente und Medikamente. Es liegen immer mehr Beweise vor, die größtenteils aus tragischen Erfahrungen stammen und belegen, dass einige Medikamente zu Störungen in der Entwicklung des Fötus führen können. Das bekannteste davon ist das Beruhigungsmittel Thalidomid, das bei vielen Kindern, deren Mütter das Medikament während der Schwangerschaft eingenommen haben, zu unterentwickelten Gliedmaßen geführt hat. Derzeit sind sich die meisten Ärzte darüber im Klaren, dass die medikamentöse Behandlung schwangerer Frauen auf ein Minimum beschränkt werden sollte, insbesondere in den ersten drei Monaten, in denen die Organbildung stattfindet. Die Einnahme jeglicher Medikamente durch eine schwangere Frau in Form von Tabletten und Kapseln sowie Hormonen und sogar Inhalationsaerosolen ist nur unter strenger Aufsicht eines Gynäkologen zulässig.
Wenn eine schwangere Frau große Mengen Alkohol trinkt, erhöht sich das Risiko, dass das Baby zahlreiche Erkrankungen entwickelt, die zusammenfassend als fetales Alkoholsyndrom bezeichnet werden. Dazu gehören Wachstumsverzögerung, geistige Behinderung, Herz-Kreislauf-Anomalien, ein kleiner Kopf (Mikrozephalie) und ein schlechter Muskeltonus.
Beobachtungen haben gezeigt, dass Kokainkonsum bei schwangeren Frauen zu schwerwiegenden Problemen beim Fötus führt. Auch andere Drogen wie Marihuana, Haschisch und Meskalin sind potenziell gefährlich. Es wurde ein Zusammenhang zwischen dem Konsum der halluzinogenen Droge LSD durch schwangere Frauen und der Häufigkeit spontaner Fehlgeburten festgestellt. Experimentellen Daten zufolge kann LSD Störungen in der Chromosomenstruktur verursachen, was auf die Möglichkeit einer genetischen Schädigung beim ungeborenen Kind hinweist (siehe LSD).
Auch das Rauchen werdender Mütter wirkt sich negativ auf den Fötus aus. Studien haben gezeigt, dass die Fälle von Frühgeburten und fetaler Unterentwicklung proportional zur Anzahl der gerauchten Zigaretten zunehmen. Rauchen kann auch die Häufigkeit von Fehl- und Totgeburten sowie die Kindersterblichkeit unmittelbar nach der Geburt erhöhen.
Strahlung. Ärzte und Wissenschaftler weisen zunehmend auf die Gefahr hin, die mit der kontinuierlichen Zunahme von Strahlungsquellen einhergeht, die Schäden am genetischen Apparat von Zellen verursachen können. In den frühen Stadien der Schwangerschaft sollten Frauen nicht unnötig Röntgenstrahlen und anderen Strahlen ausgesetzt werden. Im weiteren Sinne ist eine strenge Kontrolle medizinischer, industrieller und militärischer Strahlungsquellen von entscheidender Bedeutung für die Erhaltung der genetischen Gesundheit künftiger Generationen. Siehe auch REPRODUKTION; MENSCHLICHE REPRODUKTION; Embryologen
Http://www.krugosvet.ru/enc/medicina/EMBRIOLOGIYA_CHELOVEKA.html
Der menschliche Körper ist ein komplexes System, dessen Aktivität auf verschiedenen Ebenen reguliert wird. Gleichzeitig müssen bestimmte Stoffe an bestimmten biochemischen Prozessen beteiligt sein, damit alle Zellen, Organe und ganzen Systeme richtig funktionieren können. Und dafür müssen Sie den richtigen Grundstein legen. So wie ein mehrstöckiges Gebäude ohne ein ordnungsgemäß vorbereitetes Fundament nicht bestehen kann, erfordert der „Aufbau“ des menschlichen Körpers die korrekte Übertragung von Erbmaterial. Es ist der darin eingebettete genetische Code, der die Entwicklung des Embryos steuert, die Entstehung aller Interaktionen ermöglicht und die normale Existenz eines Menschen bestimmt.
In einigen Fällen treten jedoch Fehler in den Erbinformationen auf. Sie können auf der Ebene einzelner Gene auftreten oder deren große Assoziationen betreffen. Solche Veränderungen nennt man Genmutationen. In manchen Situationen betrifft das Problem ganze Chromosomen, also die Struktureinheiten der Zelle. Dementsprechend werden sie als Chromosomenmutationen bezeichnet. Erbkrankheiten, die als Folge von Störungen des Chromosomensatzes oder der Chromosomenstruktur entstehen, werden als chromosomal bezeichnet.
Normalerweise enthält jede Körperzelle die gleiche Anzahl an Chromosomen, gepaart mit den gleichen Genen. Beim Menschen besteht der komplette Satz aus 23 Paaren, nur in den Keimzellen ist statt 46 Chromosomen die Hälfte vorhanden. Dies ist notwendig, damit im Befruchtungsprozess, wenn Spermium und Eizelle verschmelzen, eine vollständige Kombination mit allen notwendigen Genen entsteht. Gene sind nicht zufällig, sondern in einer genau definierten Reihenfolge entlang der Chromosomen verteilt. Dabei bleibt der lineare Ablauf für alle Menschen gleich.
Bei der Bildung von Keimzellen können jedoch verschiedene „Fehler“ passieren. Durch Mutationen verändert sich die Anzahl der Chromosomen oder deren Struktur. Aus diesem Grund kann die Eizelle nach der Befruchtung einen Überschuss oder umgekehrt eine unzureichende Menge an Chromosomenmaterial enthalten. Aufgrund des Ungleichgewichts wird der Entwicklungsprozess des Embryos gestört, was zu einem Spontanabort, einer Totgeburt oder der Entwicklung einer erblichen Chromosomenerkrankung führen kann.
Ätiologie chromosomaler Erkrankungen
Zu den ätiologischen Faktoren chromosomaler Pathologien zählen alle Arten von Chromosomenmutationen. Darüber hinaus können auch einige genomische Mutationen einen ähnlichen Effekt haben.
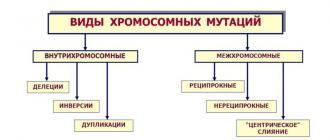
Beim Menschen kommen Deletionen, Duplikationen, Translokationen und Inversionen vor, also alle Arten von Mutationen. Bei der Löschung und Vervielfältigung liegt die genetische Information in unzureichender bzw. übermäßiger Menge vor. Da moderne Methoden das Fehlen auch nur eines kleinen Teils des genetischen Materials (auf Genebene) nachweisen können, ist es nahezu unmöglich, eine klare Grenze zwischen genetischen und chromosomalen Erkrankungen zu ziehen.
Translokationen sind der Austausch von genetischem Material, der zwischen einzelnen Chromosomen stattfindet. Mit anderen Worten: Ein Abschnitt der genetischen Sequenz wandert auf ein nicht homologes Chromosom. Unter den Translokationen werden zwei wichtige Gruppen unterschieden: reziproke und Robertsonsche.
Translokationen reziproker Natur ohne Verlust der beteiligten Bereiche werden als balanciert bezeichnet. Sie verursachen ebenso wie Inversionen keinen Verlust der Geninformation und führen daher nicht zu pathologischen Auswirkungen. Mit der weiteren Beteiligung solcher Chromosomen am Crossing-Over- und Reduktionsprozess können jedoch Gameten mit unausgeglichenen Sätzen und einem unzureichenden Satz an Genen gebildet werden. Ihre Beteiligung am Befruchtungsprozess führt zur Entwicklung bestimmter erblicher Syndrome bei den Nachkommen.
Robertsonsche Translokationen sind durch die Beteiligung zweier akrozentrischer Chromosomen gekennzeichnet. Dabei gehen die kurzen Arme verloren, während die langen erhalten bleiben. Aus 2 Ausgangschromosomen wird ein festes, metazentrisches Chromosom gebildet. Trotz des Verlusts eines Teils des genetischen Materials kommt es in diesem Fall in der Regel nicht zur Entwicklung von Pathologien, da die Funktionen der verlorenen Bereiche durch ähnliche Gene in den verbleibenden 8 akrozentrischen Chromosomen kompensiert werden.
Bei terminalen Deletionen (d. h. wenn sie verloren gehen) kann sich ein Ringchromosom bilden. Sein Träger, der solches Genmaterial von einem der Eltern erhalten hat, weist in den Endabschnitten eine partielle Monosomie auf. Wenn das Zentromer durchbrochen wird, kann ein Isochromosom gebildet werden, dessen Arme denselben Satz an Genen aufweisen (auf einem regulären Chromosom sind sie unterschiedlich).
In einigen Fällen kann sich eine uniparentale Disomie entwickeln. Sie tritt auf, wenn während der Nichtdisjunktion der Chromosomen und der Befruchtung eine Trisomie auftritt und danach eines der drei Chromosomen entfernt wird. Der Mechanismus dieses Phänomens ist derzeit nicht untersucht. Dadurch erscheinen jedoch zwei Kopien des Chromosoms eines Elternteils im Chromosomensatz, während ein Teil der genetischen Information des zweiten Elternteils verloren geht.
Die Vielfalt der Varianten der Verzerrung des Chromosomensatzes verursacht verschiedene Formen von Krankheiten.

Es gibt drei Grundprinzipien, die eine genaue Klassifizierung der resultierenden Chromosomenpathologie ermöglichen. Ihre Einhaltung liefert einen eindeutigen Hinweis auf die Form der Abweichung.
Nach dem ersten Prinzip ist es notwendig, die genetischen oder chromosomalen Merkmale der Mutation zu bestimmen und auch das spezifische Chromosom eindeutig zu identifizieren. Beispielsweise könnte es sich um eine einfache Trisomie 21 oder Triploidie handeln. Die Kombination eines einzelnen Chromosoms und der Art der Mutation bestimmt die Formen der Chromosomenpathologie. Durch die Beachtung dieses Prinzips ist es möglich, genau zu bestimmen, in welcher Struktureinheit Veränderungen vorliegen, und auch herauszufinden, ob ein Überschuss oder ein Mangel an Chromosomenmaterial festgestellt wurde. Dieser Ansatz ist effektiver als die Klassifizierung nach klinischen Symptomen, da viele Abweichungen ähnliche Entwicklungsstörungen des Körpers verursachen.
Nach dem zweiten Prinzip ist es notwendig, den Zelltyp zu bestimmen, in dem die Mutation aufgetreten ist – Zygote oder Gamete. Mutationen in Gameten führen zum Auftreten vollständiger Formen von Chromosomenerkrankungen. Jede Zelle im Körper enthält eine Kopie des genetischen Materials mit der Chromosomenanomalie. Wenn die Störung später auftritt, im Zygotenstadium oder während der Spaltung, wird die Mutation als somatisch eingestuft. Dabei erhalten einige der Zellen das ursprüngliche genetische Material, andere einen veränderten Chromosomensatz. Im Körper können zwei oder mehr Arten von Sets gleichzeitig vorhanden sein. Ihre Kombination ähnelt einem Mosaik, weshalb diese Form der Krankheit als Mosaik bezeichnet wird. Wenn im Körper mehr als 10 % Zellen mit verändertem Chromosomensatz vorhanden sind, wiederholt sich das Krankheitsbild in voller Form.
Nach dem dritten Prinzip wird die Generation identifiziert, in der die Mutation erstmals aufgetreten ist. Wurde bei gesunden Eltern eine Veränderung der Gameten festgestellt, spricht man von einem sporadischen Fall. War es bereits im mütterlichen oder väterlichen Körper vorhanden, handelt es sich um eine vererbte Form. Ein erheblicher Anteil der vererbten Chromosomenerkrankungen wird durch Robertson-Translokationen, Inversionen und balancierte reziproke Translokationen verursacht. Im Verlauf der Meiose können sie zur Bildung einer pathologischen Kombination führen.
Eine vollständig genaue Diagnose setzt voraus, dass die Art der Mutation, das betroffene Chromosom festgestellt, der vollständige oder mosaikartige Charakter der Krankheit geklärt wurde und eine erbliche Übertragung oder ein sporadisches Auftreten festgestellt wurde. Die hierfür notwendigen Daten können durch eine genetische Diagnostik anhand von Proben des Patienten und in manchen Fällen auch seiner Angehörigen gewonnen werden.
Allgemeine Probleme
Die intensive Entwicklung der Genetik in den letzten Jahrzehnten hat es ermöglicht, einen eigenen Bereich der Chromosomenpathologie zu entwickeln, der nach und nach immer mehr an Bedeutung gewinnt. In diesen Bereich fallen nicht nur Chromosomenerkrankungen, sondern auch verschiedene Störungen während der intrauterinen Entwicklung (z. B. Fehlgeburten). Derzeit beträgt die Zahl der Anomalien 1000. Über hundert Formen zeichnen sich durch ein klinisch definiertes Bild aus und werden als Syndrome bezeichnet.
Es gibt mehrere Gruppen von Krankheiten. Triploidie ist eine Erkrankung, bei der die Körperzellen über eine zusätzliche Kopie des Genoms verfügen. Tritt ein Duplikat nur eines Chromosoms auf, spricht man von Trisomie. Die Ursachen für eine abnormale Entwicklung des Körpers können auch Deletionen (entfernte Abschnitte des genetischen Codes), Duplikationen (bzw. zusätzliche Kopien von Genen oder ihrer Gruppen) und andere Defekte sein. Der englische Arzt L. Down beschrieb 1866 eine der bekanntesten Krankheiten dieser Art. Das nach ihm benannte Syndrom tritt auf, wenn eine zusätzliche Kopie des Chromosoms 21 vorhanden ist (Trisomie 21). Trisomien auf anderen Chromosomen führen aufgrund schwerwiegender Entwicklungsstörungen meist zu Fehlgeburten oder zum Tod im Kindesalter.
Später wurden Fälle von Monosomie auf dem X-Chromosom entdeckt. Im Jahr 1925 beschrieben N. A. Shereshevsky und G. Turner 1938 seine Symptome. Trisomie-XXY, die bei Männern auftritt, wurde 1942 von Klinefelter beschrieben.
Diese Krankheitsfälle wurden zu den ersten Forschungsobjekten auf diesem Gebiet. Nachdem die Ätiologie der drei aufgeführten Syndrome entschlüsselt war, zeigte sich tatsächlich die Richtung der Chromosomenerkrankungen. In den 1960er Jahren führten weitere zytogenetische Studien zur Entstehung der klinischen Zytogenetik. Wissenschaftler haben den Zusammenhang zwischen pathologischen Anomalien und chromosomalen Mutationen nachgewiesen und auch statistische Daten zur Häufigkeit von Mutationen bei Neugeborenen und bei Spontanaborten erhalten.
Arten von Chromosomenanomalien
Chromosomenanomalien können entweder relativ groß oder klein sein. Je nach Größe ändern sich die Forschungsmethoden. Beispielsweise können Punktmutationen, Deletionen und Duplikationen in Regionen mit einer Länge von hundert Nukleotiden nicht mit dem Mikroskop nachgewiesen werden. Die Bestimmung einer Chromosomenstörung mit der Methode der Differentialfärbung ist nur möglich, wenn die Größe des betroffenen Bereichs in Millionen Nukleotiden berechnet wird. Kleinere Mutationen können nur durch die Bestimmung der Nukleotidsequenz identifiziert werden. In der Regel führen größere Störungen (z. B. unter dem Mikroskop sichtbar) zu einer stärkeren Beeinträchtigung der Körperfunktionen. Darüber hinaus kann die Anomalie nicht nur das Gen betreffen, sondern auch einen Teil des Erbmaterials, dessen Funktionen derzeit nicht untersucht werden.
Monosomie ist eine Anomalie, die zum Fehlen eines der Chromosomen führt. Der umgekehrte Fall ist Trisomie – die Hinzufügung einer zusätzlichen Kopie eines Chromosoms zum Standardsatz von 23 Paaren. Dementsprechend ändert sich auch die Anzahl der Kopien von Genen, die normalerweise in zwei Kopien vorliegen. Bei der Monosomie fehlt das Gen, bei der Trisomie ist es im Überschuss vorhanden. Führt eine Chromosomenanomalie zu einer Veränderung der Anzahl einzelner Abschnitte, spricht man von partieller Trisomie oder Monosomie (zum Beispiel am Arm 13q).
Es gibt auch Fälle von uniparentaler Disomie. In diesem Fall gelangt ein Paar homologer Chromosomen (oder ein dazu homologer Teil) von einem der Elternteile in den Körper. Der Grund ist ein unerforschter Mechanismus, der vermutlich aus zwei Phasen besteht – der Bildung einer Trisomie und der Entfernung eines der drei Chromosomen. Die Auswirkungen einer uniparentalen Disomie können geringfügig oder erheblich sein. Tatsache ist, dass sich ein rezessiv mutiertes Allel auf identischen Chromosomen automatisch manifestiert. Gleichzeitig darf der Elternteil, von dem das Chromosom mit der Mutation stammt, aufgrund der Heterozygotie des Gens keine gesundheitlichen Probleme haben.
Aufgrund der hohen Bedeutung des genetischen Materials für alle Entwicklungsstadien eines Organismus können bereits kleine Anomalien große Veränderungen in der koordinierten Aktivität von Genen verursachen. Schließlich wurde ihre gemeinsame Arbeit über Millionen von Jahren der Evolution verfeinert. Es ist nicht überraschend, dass sich die Folgen einer solchen Mutation höchstwahrscheinlich auf Gametenebene manifestieren. Sie wirken sich besonders stark auf Männer aus, da der Embryo irgendwann vom weiblichen auf den männlichen Entwicklungsweg wechseln muss. Bei unzureichender Aktivität der entsprechenden Gene kommt es zu verschiedenen Abweichungen, bis hin zum Hermaphroditismus.

Die ersten Studien zu den Auswirkungen von Chromosomenstörungen begannen in den 60er Jahren, nachdem die chromosomale Natur einiger Krankheiten festgestellt wurde. Man kann grob zwei große Gruppen assoziierter Auswirkungen unterscheiden: angeborene Fehlbildungen und Veränderungen, die zum Tod führen. Die moderne Wissenschaft hat Informationen darüber, dass Chromosomenanomalien bereits im Zygotenstadium auftreten. Tödliche Wirkungen sind in diesem Fall eine der Hauptursachen für den Tod des Fötus im Mutterleib (dieser Indikator ist beim Menschen ziemlich hoch).
Chromosomenaberrationen sind Veränderungen in der Struktur des Chromosomenmaterials. Sie können sporadisch auftreten oder vererbt werden. Der genaue Grund für ihr Erscheinen ist nicht geklärt. Wissenschaftler glauben, dass einige dieser Mutationen für verschiedene Umweltfaktoren (zum Beispiel chemisch aktive Substanzen) verantwortlich sind, die sich auf den Embryo oder sogar die Zygote auswirken. Eine interessante Tatsache ist, dass die meisten Chromosomenaberrationen normalerweise mit den Chromosomen zusammenhängen, die der Embryo vom Vater erhält.
Ein erheblicher Teil der Chromosomenaberrationen ist sehr selten und wurde nur einmal entdeckt. Gleichzeitig kommen einige andere durchaus häufig vor, auch bei Menschen, die nicht verwandt sind. Beispielsweise ist die Translokation zentromerer oder benachbarter Regionen der Chromosomen 13 und 14 weit verbreitet. Der Verlust von inaktivem Kurzarmchromatin hat praktisch keine Auswirkungen auf die Gesundheit. Bei ähnlichen Robertson-Translokationen landen 45 Chromosomen im Karyotyp.
Ungefähr zwei Drittel aller bei Neugeborenen festgestellten Chromosomenanomalien werden durch andere Genkopien ausgeglichen. Aus diesem Grund stellen sie keine ernsthafte Gefahr für die normale Entwicklung des Kindes dar. Ist eine Kompensation der Störung nicht möglich, kommt es zu Entwicklungsstörungen. Eine solche unausgeglichene Anomalie wird häufig bei Patienten mit geistiger Behinderung und anderen angeborenen Defekten sowie bei Feten nach spontanen Abtreibungen festgestellt.
Es sind kompensierte Anomalien bekannt, die von Generation zu Generation vererbt werden können, ohne dass Krankheiten auftreten. In einigen Fällen kann sich eine solche Anomalie zu einer unausgeglichenen Form entwickeln. Liegt also eine Translokation des Chromosoms 21 vor, steigt das Risiko einer Trisomie. Laut Statistik hat jedes 20. Kind mit Trisomie-21 eine solche Translokation, und in jedem fünften Fall hat ein Elternteil eine ähnliche Störung. Da die meisten Kinder mit einer durch Translokation verursachten Trisomie 21 von jungen Müttern (unter 30 Jahren) geboren werden, ist beim Nachweis dieser Erkrankung bei einem Kind eine diagnostische Untersuchung der jungen Eltern erforderlich.
Das Risiko des Auftretens nicht kompensierter Störungen hängt stark von der Translokation ab, sodass theoretische Berechnungen schwierig sind. Die Wahrscheinlichkeit der entsprechenden Pathologie kann jedoch anhand statistischer Daten näherungsweise bestimmt werden. Solche Informationen werden für häufige Translokationen gesammelt. Insbesondere eine Robertsonsche Translokation zwischen den Chromosomen 14 und 21 bei der Mutter hat eine Wahrscheinlichkeit von 2 Prozent, beim Kind zu Trisomie 21 zu führen. Die gleiche Translokation beim Vater wird mit einer Wahrscheinlichkeit von 10 % vererbt.
Prävalenz von Chromosomenanomalien
Forschungsergebnisse zeigen, dass mindestens ein Zehntel der Eizellen nach der Befruchtung und etwa 5–6 Prozent der Föten verschiedene Chromosomenanomalien aufweisen. In der Regel kommt es in diesem Fall nach 8–11 Wochen zu einem Spontanabort. In einigen Fällen führen sie später zu Fehlgeburten oder führen zu einer Totgeburt.
Bei Neugeborenen (basierend auf einer Befragung von mehr als 65.000 Kindern) treten bei etwa 0,5 % der Gesamtzahl Veränderungen in der Chromosomenzahl oder signifikante Chromosomenaberrationen auf. Mindestens einer von 700 leidet an Trisomie 13, 18 oder 21; Etwa 1 von 350 Jungen hat einen auf 47 Einheiten erweiterten Chromosomensatz (Karyotypen 47,XYY und 47,XXY). Monosomie auf dem X-Chromosom ist seltener – Einzelfälle von mehreren Tausend. Etwa 0,2 % weisen kompensierte Chromosomenaberrationen auf.
Bei Erwachsenen werden manchmal auch erbliche Anomalien (meist kompensiert) festgestellt, manchmal mit Trisomie der Geschlechtschromosomen. Untersuchungen zeigen auch, dass etwa 10–15 Prozent aller Fälle geistiger Behinderung durch das Vorliegen einer Chromosomenanomalie erklärt werden können. Dieser Wert erhöht sich deutlich, wenn neben psychischen Entwicklungsstörungen auch anatomische Defekte beobachtet werden. Unfruchtbarkeit wird häufig auch durch ein zusätzliches Geschlechtschromosom (bei Männern) und eine Monosomie/Aberration auf dem X-Chromosom (bei Frauen) verursacht.
Zusammenhang zwischen Chromosomenanomalien und bösartigen Erkrankungen
In der Regel führt die Untersuchung bösartiger Tumorzellen zum Nachweis mikroskopisch sichtbarer Chromosomenanomalien. Tests auf Leukämie, Lymphome und eine Reihe anderer Krankheiten liefern ähnliche Ergebnisse.
Insbesondere bei Lymphomen ist es nicht ungewöhnlich, eine Translokation zu finden, die mit einem Bruch innerhalb oder in der Nähe des Immunglobulin-Schwerkette-Locus (Chromosom 14) einhergeht. In diesem Fall wandert das MYC-Gen von Chromosom 8 auf 14.
Bei myeloischer Leukämie wird in den meisten Fällen (über 95 %) eine Translokation zwischen den Chromosomen 22 und 9 festgestellt, die zum Auftreten des charakteristischen „Philadelphia“-Chromosoms führt.
Während der Entwicklung geht die Explosionskrise mit dem Auftreten aufeinanderfolgender Chromosomenanomalien im Karyotyp einher.
Durch differenzielle Färbeverfahren mit anschließender Beobachtung unter dem Mikroskop sowie molekulargenetische Untersuchungsmethoden ist es möglich, Chromosomenanomalien bei verschiedenen Leukämien frühzeitig zu erkennen. Diese Informationen helfen, eine Entwicklungsprognose zu erstellen, die Diagnose zu klären und die Therapie anzupassen.
Bei häufigen soliden Tumoren wie Darmkrebs, Brustkrebs usw. Konventionelle zytogenetische Methoden sind mit einigen Einschränkungen anwendbar. Es wurden jedoch auch charakteristische Chromosomenanomalien festgestellt. Bei Tumoren gefundene Anomalien hängen oft mit Genen zusammen, die für den Prozess des normalen Zellwachstums verantwortlich sind. Aufgrund der Amplifikation (Bildung mehrerer Kopien) des Gens wird manchmal die Bildung kleiner Minichromosomen in Neoplasmazellen beobachtet.
In einigen Fällen wird das Auftreten eines bösartigen Tumors durch den Verlust eines Gens verursacht, das die Proliferation unterdrücken soll. Dafür kann es mehrere Gründe geben: Löschungen und Brüche während des Translokationsprozesses sind am häufigsten. Mutationen dieser Art gelten im Allgemeinen als rezessiv, da das Vorhandensein auch nur eines normalen Allels normalerweise eine ausreichende Wachstumskontrolle gewährleistet. Störungen können auftreten oder vererbt werden. Wenn im Genom keine normale Kopie des Gens vorhanden ist, hängt die Proliferation nicht mehr von regulatorischen Faktoren ab.
Daher sind die folgenden Arten die wichtigsten Chromosomenanomalien, die das Auftreten und Wachstum bösartiger Neubildungen beeinflussen:
Translokationen, da sie zu einer Störung der normalen Funktion der für die Proliferation verantwortlichen Gene führen (oder deren erhöhte Arbeit verursachen können);
Deletionen, die zusammen mit anderen rezessiven Mutationen Veränderungen in der Regulierung des Zellwachstums verursachen;
Rezessive Mutationen werden durch Rekombination homozygot und manifestieren sich daher vollständig.
Verstärkungen, die die Tumorzellproliferation stimulieren.
Die Identifizierung dieser Mutationen im Rahmen der genetischen Diagnose kann auf ein erhöhtes Risiko für die Entwicklung bösartiger Neubildungen hinweisen.
Bekannte Erkrankungen chromosomaler Natur
Eine der bekanntesten Krankheiten, die aufgrund von Anomalien im Erbgut entsteht, ist das Down-Syndrom. Es wird durch Trisomie 21-Chromosomen verursacht. Ein charakteristisches Symptom dieser Krankheit ist eine Entwicklungsverzögerung. Kinder haben beim Lernen in der Schule ernsthafte Probleme und benötigen oft eine alternative Methode zum Unterrichten des Stoffes. Gleichzeitig werden körperliche Entwicklungsstörungen festgestellt – ein flaches Gesicht, vergrößerte Augen, Klinodaktylie und andere. Wenn solche Menschen große Anstrengungen unternehmen, können sie recht gut Kontakte knüpfen; es gibt sogar einen bekannten Fall, in dem ein Mann mit Down-Syndrom erfolgreich eine höhere Ausbildung erlangte. Patienten haben ein erhöhtes Risiko, an Demenz zu erkranken. Dies und eine Reihe anderer Gründe führen zu einer kurzen Lebenserwartung.
Zur Trisomie gehört auch das Patau-Syndrom, nur dass in diesem Fall drei Kopien des Chromosoms 13 vorhanden sind. Die Krankheit ist durch multiple Entwicklungsstörungen gekennzeichnet, häufig mit Polydaktylie. In den meisten Fällen liegt eine Störung der Aktivität des Zentralnervensystems oder dessen Unterentwicklung vor. Häufig (etwa 80 Prozent) der Patienten haben Herzfehler. Schwere Erkrankungen führen zu einer hohen Sterblichkeit – bis zu 95 % der Kinder mit dieser Diagnose sterben im ersten Lebensjahr. Eine Behandlung oder Korrektur der Erkrankung ist in der Regel nicht möglich, lediglich eine einigermaßen ständige Überwachung des Zustandes des Betroffenen ist möglich.
Eine weitere Form der Trisomie, mit der Kinder geboren werden, liegt auf dem Chromosom 18. Die Krankheit wird in diesem Fall Edwards-Syndrom genannt und ist durch mehrere Störungen gekennzeichnet. Knochen sind deformiert, häufig wird auch eine veränderte Form des Schädels beobachtet. Das Herz-Kreislauf-System weist meist Entwicklungsstörungen auf, auch am Atmungssystem werden Probleme festgestellt. Dies hat zur Folge, dass etwa 60 % der Kinder im Alter von 1 Jahr nicht mehr leben; bis zu 95 % der Kinder mit dieser Diagnose sterben.
Eine Trisomie auf anderen Chromosomen kommt bei Neugeborenen praktisch nie vor, da sie fast immer zu einem vorzeitigen Schwangerschaftsabbruch führt. In einigen Fällen kommt es zu einer Totgeburt des Babys.
Das Shereshevsky-Turner-Syndrom geht mit Störungen der Anzahl der Geschlechtschromosomen einher. Aufgrund von Störungen im Prozess der Chromosomentrennung geht das X-Chromosom im weiblichen Körper verloren. Dadurch erhält der Körper nicht die richtige Menge an Hormonen, sodass seine Entwicklung gestört ist. Dies gilt vor allem für die Genitalien, die sich nur teilweise entwickeln. Für eine Frau bedeutet dies fast immer die Unfähigkeit, Kinder zu bekommen.
Bei Männern führt eine Polysomie auf dem Y- oder X-Chromosom zur Entwicklung des Klinefelter-Syndroms. Diese Krankheit ist durch eine schwache Ausprägung männlicher Merkmale gekennzeichnet. Oft begleitet von einer Gynäkomastie sind Entwicklungsverzögerungen möglich. In den meisten Fällen werden frühe Potenzprobleme und Unfruchtbarkeit beobachtet. In diesem Fall kann wie beim Shereshevsky-Turner-Syndrom eine In-vitro-Fertilisation eine Lösung sein.
Dank pränataldiagnostischer Methoden ist es möglich, diese und andere Erkrankungen des Fötus während der Schwangerschaft zu erkennen. Paare können sich dazu entschließen, eine Schwangerschaft abzubrechen, um zu versuchen, ein weiteres Kind zu bekommen. Wenn sie sich entscheiden, ein Kind auszutragen und zur Welt zu bringen, können sie sich durch die Kenntnis der Eigenschaften seines genetischen Materials im Voraus auf bestimmte Methoden der Vorbeugung oder Behandlung vorbereiten.
Ein Karyotyp ist ein systematischer Satz von Chromosomen eines Zellkerns mit seinen quantitativen und qualitativen Merkmalen.

Normaler weiblicher Karyotyp – 46.XX Normaler männlicher Karyotyp – 46.XY
Die Karyotyp-Untersuchung ist ein Verfahren zur Identifizierung von Abweichungen in der Struktur und Anzahl der Chromosomen.
Indikationen zur Karyotypisierung:
- Mehrere angeborene Fehlbildungen, begleitet von einem klinisch abnormalen Phänotyp oder Dysmorphismus
- Geistige Behinderung oder Entwicklungsverzögerung
- Störung der sexuellen Differenzierung oder Anomalien der sexuellen Entwicklung
- Primäre oder sekundäre Amenorrhoe
- Anomalien im Spermogramm – Azoospermie oder schwere Oligospermie
- Unfruchtbarkeit unbekannter Ätiologie
- Gewohnheitsmäßige Fehlgeburt
- Eltern eines Patienten mit strukturellen Chromosomenanomalien
- Wiederholte Geburt von Kindern mit Chromosomenanomalien
Leider können Karyotypstudien nur große strukturelle Veränderungen feststellen. In den meisten Fällen handelt es sich bei Anomalien in der Chromosomenstruktur um Mikrodeletionen und Mikroduplikationen, die unter dem Mikroskop nicht sichtbar sind. Solche Veränderungen lassen sich jedoch mit modernen molekularzytogenetischen Methoden – Fluoreszenzhybridisierung (FISH) und chromosomaler Microarray-Analyse – gut identifizieren.
Die Abkürzung FISH steht für Fluoreszenz-in-situ-Hybridisierung. Hierbei handelt es sich um eine zytogenetische Methode, mit der die Position einer bestimmten DNA-Sequenz auf Chromosomen identifiziert und bestimmt wird. Zu diesem Zweck werden spezielle Sonden verwendet – Nukleoside, die mit Fluorophoren oder anderen Markierungen verbunden sind. Die Visualisierung gebundener DNA-Sonden erfolgt mit einem Fluoreszenzmikroskop.
Die FISH-Methode ermöglicht die Untersuchung kleiner Chromosomenumlagerungen, die durch Standard-Karyotyptests nicht identifiziert werden. Es hat jedoch einen wesentlichen Nachteil. Sonden sind nur für eine Region des Genoms spezifisch und daher ist es in einer Studie möglich, das Vorhandensein oder die Anzahl der Kopien nur dieser Region (oder mehrerer bei Verwendung mehrfarbiger Sonden) zu bestimmen. Daher ist der korrekte klinische Hintergrund wichtig und die FISH-Analyse kann die Diagnose nur bestätigen oder nicht bestätigen.
Eine Alternative zu dieser Methode ist die chromosomale Microarray-Analyse, die mit der gleichen Genauigkeit, Sensitivität und Spezifität die Menge des genetischen Materials an Hunderttausenden (und sogar Millionen) Punkten im Genom bestimmt, was eine Diagnose fast aller ermöglicht bekannte Mikrodeletions- und Mikroduplikationssyndrome.

Die chromosomale Microarray-Analyse ist eine molekularzytogenetische Methode zur Identifizierung von Variationen in der DNA-Kopienzahl im Vergleich zu einer Kontrollprobe. Bei der Durchführung dieser Analyse werden alle klinisch bedeutsamen Regionen des Genoms untersucht, wodurch eine chromosomale Pathologie des Patienten mit höchster Genauigkeit ausgeschlossen werden kann. Auf diese Weise können pathogene Deletionen (Verschwinden von Chromosomenabschnitten), Duplikationen (Auftauchen zusätzlicher Kopien des genetischen Materials), Bereiche mit Verlust der Heterozygotie, die für Prägungskrankheiten wichtig sind, blutsverwandte Ehen und autosomal rezessive Erkrankungen identifiziert werden.
Wann ist eine chromosomale Microarray-Analyse notwendig?
- Als Erstlinientest zur Diagnose von Patienten mit Dysmorphie, angeborenen Fehlbildungen, geistiger Behinderung/Entwicklungsverzögerung, mehreren angeborenen Anomalien, Autismus, Krampfanfällen oder einem vermuteten genomischen Ungleichgewicht.
- Als Ersatz für Karyotyp, FISH und vergleichende genomische Hybridisierung bei Verdacht auf ein Mikrodeletions-/Mikroduplikationssyndrom.
- Als Test zur Erkennung unausgeglichener Chromosomenaberrationen.
- Als zusätzlicher diagnostischer Test für monogene Erkrankungen, die mit dem Funktionsverlust eines Allels einhergehen (Haploinsuffizienz), insbesondere wenn die Sequenzierung keine pathogene Mutation identifizieren kann und die Deletion des gesamten Gens die Ursache sein kann.
- Bestimmung der Herkunft des genetischen Materials bei uniparentalen Disomien, Duplikationen und Deletionen.
1 Test – 400 Syndrome (Liste)
Einführung in die Chromosomen-Microarray-Analyse.
Informationen für Ärzte
Regeln für das Sammeln von Material für die chromosomale Microarray-Analyse
Der Inhalt des Artikels
Angeborene Defekte, Störungen in der Struktur, den Funktionen und der Biochemie des Körpers, die durch angeborene oder pränatale Ursachen verursacht werden und zu körperlichen oder geistigen Beeinträchtigungen, Krankheiten oder zum Tod führen. Zu den pränatalen Ursachen solcher Defekte zählen erbliche Faktoren und (oder) Umwelteinflüsse auf die Entwicklung des Embryos. Die Ursache für Fehlbildungen während der Geburt können Verletzungen oder Infektionen sein. Ein sehr niedriges Geburtsgewicht, das entweder auf eine Frühgeburt oder ein Versagen der fetalen Entwicklung zurückzuführen ist und eine der Hauptursachen für Säuglingssterblichkeit und Behinderung darstellt, gilt ebenfalls als Geburtsfehler.
Historischer Aspekt.
Prähistorische Kunst zeigt, dass Geburtsfehler seit der Antike bekannt sind. Ihr Aussehen löste Angst aus und ließ viele Mythen entstehen. Keilschrifttafeln aus dem alten Babylon berichten, dass angeborene Missbildungen als Omen von nationaler Bedeutung galten und als Warnungen wütender Götter entziffert wurden. Es herrschte die weitverbreitete Überzeugung, dass die Eindrücke der Mutter während der Schwangerschaft Einfluss auf die Bildung des Kindes hätten; Sie dachten, dass eine gespaltene Lippe (die sogenannte „Spalte“) die Folge der Angst vor einem Hasen sei, und dass deformierte Beine nach einer Begegnung mit einem Krüppel auftraten. Andere Glaubensrichtungen waren die Ursache für das Leiden und den Tod von Mutter und Kind, da sie beispielsweise behaupteten, dass ein monströser Nachwuchs das Ergebnis einer fleischlichen Beziehung zu einem Tier sei.
Eine der ersten Beobachtungen, die die Natur angeborener Defekte aufdeckten, stammt aus dem Jahr 1651 und gehört dem englischen Arzt William Harvey. Er stellte fest, dass einige der Defekte das Ergebnis der Erhaltung eines für den Embryo (oder Fötus) normalen Merkmals sind, das normalerweise zum Zeitpunkt der Geburt verschwindet. Allerdings erst im 19. Jahrhundert. Entwicklungsdefekte wurden im 20. Jahrhundert sorgfältig untersucht. war geprägt von der Entwicklung der Genforschung, und die gewonnenen Erkenntnisse ersetzten den phantastischen, oft schädlichen Aberglauben der Vergangenheit; Zum ersten Mal wurden Methoden zur Vorbeugung und Behandlung einiger dieser schweren Erkrankungen entwickelt.
URSACHEN FÜR GEBURTSFEHLER
Vererbung.
Einige Geburtsfehler werden auf die gleiche Weise vererbt wie andere Merkmale. Erbliche Informationen werden mithilfe von Genen, deren Träger Chromosomen sind, von den Eltern auf die Kinder übertragen. Normalerweise enthält jede Geschlechtszelle (Sperma oder Ei) 23 Chromosomen. Während der Befruchtung, d.h. Durch die Verschmelzung von Spermium und Eizelle wird der normale genetische Satz von 46 Chromosomen wiederhergestellt. 22 der 23 Chromosomen einer Fortpflanzungszelle sind Autosomen, d. h. Sie bestimmen nicht das Geschlecht, aber eines davon ist entweder das X- oder das Y-Geschlechtschromosom. Das Sperma trägt entweder das X- oder Y-Chromosom, die Eizelle trägt nur das X-Chromosom. Die Befruchtung einer Eizelle durch ein Spermium mit einem Y-Chromosom bringt einen männlichen Nachwuchs hervor, und ein Spermium mit einem X-Chromosom bringt einen weiblichen Nachwuchs hervor.
Viele erbliche Merkmale und ihre Störungen entsprechen statistisch vorhersehbaren Vererbungsmustern, die nach ihrem Entdecker Gregor Mendel Mendelsch genannt werden. Die Mendelsche Vererbung ist die am besten verstandene Methode der genetischen Übertragung von Geburtsfehlern. Letzteres kann entweder durch dominante oder rezessive Vererbung übertragen werden.
Der Genotyp jedes Elternteils trägt zwei Varianten (Allele) des Gens, das dieses Merkmal bestimmt, und das Kind erhält von jedem Elternteil ein Allel. Die Manifestation eines abnormalen Merkmals als dominantes Merkmal tritt auf, wenn ein Kind von einem Elternteil ein defektes Gen erbt, das die normale Variante des anderen Elternteils dominiert. Ein Elternteil mit einem solchen dominanten Gen hat immer eine entsprechende Störung (wenn auch möglicherweise in milder Form). Die Wahrscheinlichkeit, dass ein Kind an dieser Krankheit erkrankt, liegt bei 50 %, je nachdem, ob ihm das normale oder defekte Gen von einem erkrankten Elternteil vererbt wird. Beispiele für einen dominanten Erbgang sind Morbus Gettington (eine fortschreitende Erkrankung des Zentralnervensystems) und achondroplastischer Zwergwuchs (verzögertes Knochenwachstum).
Die Vererbung eines rezessiven Merkmals führt zu einer ausgeprägten Störung des Kindes, wenn beide Elternteile das gleiche defekte Gen (zusammen mit dem normalen Gen für dieses Merkmal) tragen, sie jedoch keine klinische Manifestation der Krankheit aufweisen. Jedes geborene Kind hat eine 25-prozentige Chance, das defekte Gen von keinem Elternteil zu erben, eine 50-prozentige Chance, Träger zu sein (nur ein defektes Gen zu haben) und eine 25-prozentige Chance, es in einer „doppelten Dosis“ zu erben ( zwei defekte Gene), wodurch die Krankheit vererbt wird. Sichelzellenanämie, verursacht durch einen Defekt im Hämoglobinmolekül (cm. SICHELZELLENANÄMIE) , - ein Beispiel für eine rezessiv vererbte Krankheit. Weitere Beispiele sind Thalassämie (eine andere Form der Anämie, die hauptsächlich bei Menschen mediterraner und asiatischer Abstammung auftritt) sowie die Tay-Sachs-Krankheit, eine Stoffwechselstörung, die im frühen Kindesalter zum Tod führt und sich hauptsächlich in Familien jüdischer Herkunft manifestiert Osteuropa.
Störungen wie die oben besprochenen werden durch ein autosomales Gen verursacht (das sich nicht auf den Geschlechtschromosomen befindet) und werden daher autosomale Erkrankungen genannt. Eine weitere Gruppe besteht aus den sogenannten. X-chromosomale oder geschlechtsgebundene Störungen; Sie werden durch ein defektes Gen auf dem X-Chromosom bestimmt. Da Frauen normalerweise zwei X-Chromosomen haben, kann eine Mutter Trägerin eines defekten X-chromosomal-rezessiven Gens sein und trotzdem gesund sein. Männer haben nur ein X-Chromosom und weisen aufgrund des Fehlens eines zweiten X-Chromosoms mit seiner kompensierenden Wirkung fast immer die Wirkung des defekten Gens auf. Jedes Kind hat eine 50-prozentige Chance, das defekte Gen von seiner Trägermutter zu erben. Frauen, die ein solches Gen erben, werden zu Trägerinnen, und Männer entwickeln die Krankheit. Ein kranker Vater kann das defekte Gen nicht an seine Söhne weitergeben, da diese sein Y-Chromosom erben, aber alle Töchter, die sein X-Chromosom erhalten, sind Träger. Farbenblindheit und Hämophilie (eine Blutgerinnungsstörung) sind X-chromosomal-rezessiv vererbte Erkrankungen. Eine andere X-chromosomale Störung, das sogenannte Fragile-X-Syndrom, führt zu geistiger Behinderung unterschiedlichen Ausmaßes. Männer sind häufiger und in schwererer Form betroffen.
Genetisch bedingte angeborene Defekte entstehen zufällig durch Genmutationen oder Fehler bei der Chromosomenreplikation während der Reifung einer Eizelle oder eines Spermiums. Die direkte Folge von Mutationen sind molekulare, qualitative und quantitative Veränderungen im Genprodukt. Gelegentlich gibt es vorteilhafte Mutationen, die meisten davon sind jedoch schädlich. Eine große Anzahl von Fällen X-chromosomaler und dominanter Erkrankungen entstehen durch neue Mutationen. Zwei bekannte Mutationsquellen sind ionisierende Strahlung und eine Reihe von Chemikalien. Bei der Entwicklung von Spermium und Eizelle müssen die Chromosomen sehr genau verdoppelt (verdoppelt) und dann verteilt werden, sodass jede reife Zelle nur die Hälfte des normalen Chromosomensatzes erhält. Aus unklaren Gründen treten jedoch bei der Divergenz der Chromosomen manchmal Fehler auf, die dazu führen können, dass einer reifen Keimzelle entweder ein Chromosom fehlt oder sie über ein zusätzliches Chromosom verfügt. Darüber hinaus können Chromosomen ungenau dupliziert oder gebrochen sein. Signifikante Chromosomenanomalien führen in der Regel zu mehreren Anomalien, die für den Embryo, den Fötus oder das Neugeborene tödlich sind, und treten insbesondere bei etwa 50 % der Fehlgeburten auf. Eine Chromosomenanomalie liegt einem der häufigsten Geburtsfehler zugrunde, dem Down-Syndrom, das durch das Vorhandensein eines zusätzlichen 21. Chromosoms verursacht wird und sich in geistiger und körperlicher Behinderung sowie einer Reihe anderer Anzeichen äußert (cm. DOWN-SYNDROM).
Die zweithäufigste Ursache für eine angeborene geistige Behinderung ist eine Chromosomenanomalie, die als fragiles X bekannt ist. Ein Defekt in der Struktur solcher X-Chromosomen liegt am Ende des langen Arms, der die Form eines Stiels mit einer tropfenförmigen Verdickung aufweist; Ein dünner Stiel bricht bei der Vorbereitung für die Mikroskopie häufig ab und wird daher als instabile Stelle (Site) bezeichnet, und das Chromosom selbst wird als fragile (fragile) bezeichnet. Es ist nicht bekannt, wie das fragile Chromosom an der Entstehung pathologischer Symptome beteiligt ist, es wurde jedoch gezeigt, dass in seiner instabilen Region eine bestimmte Sequenz von DNA-Basen (Cytosin-Guanin-Guanin) häufiger wiederholt wird. Die Bedeutung solcher Wiederholungen ist unklar.
Das Fragile-X-Syndrom wird rezessiv vererbt, d. h. Seine Wirkung kann durch das Vorhandensein eines normalen X-Chromosoms blockiert oder maskiert werden. Da Männer nur ein X-Chromosom haben, manifestiert sich das fragile X-Syndrom in vollem Umfang – mit geistiger Behinderung, vergrößerten Hoden, abstehenden Ohren und einem hervorstehenden Kinn. Frauen mit ihren beiden X-Chromosomen sollten durch das Vorhandensein eines fragilen Chromosoms nicht beeinträchtigt werden, aber überraschenderweise zeigen etwa ein Drittel der Frauen, die das defekte Chromosom tragen, eine gewisse geistige Behinderung. Aber selbst wenn sie über eine normale Intelligenz verfügen, besteht bei weiblichen Trägern eine 50-prozentige Chance, das defekte Chromosom an jedes ihrer Kinder weiterzugeben.
Es gibt Fälle, in denen embryonale Zellen nur ein X-Chromosom und kein Y-Chromosom haben; Das Ergebnis ist ein weibliches Kind mit Turner-Syndrom. In anderen Fällen enthält die befruchtete Eizelle (Zygote) neben einem Y-Chromosom ein (oder mehrere) zusätzliche X-Chromosomen; Dies führt zur Geburt eines männlichen Kindes mit Klinefelter-Syndrom. Solche Chromosomenanomalien sind durch sexuelle Unterentwicklung, Sterilität, Störungen von Entwicklungs- und Wachstumsprozessen und manchmal auch geistige Behinderung gekennzeichnet.
Gelegentlich erscheint ein zusätzliches Chromosom nicht im Sperma oder in der Eizelle, sondern im Embryo in einem frühen Stadium seiner Entwicklung – als Folge einer falschen Divergenz eines Chromosomenpaares während der Zellteilung. Alle Zellen, die von der resultierenden defekten Zelle abstammen, haben ein zusätzliches Chromosom, und das Ausmaß, in dem der Defekt ein Individuum betrifft, hängt weitgehend davon ab, wie früh in der Entwicklung der Fehler aufgetreten ist. Diese Abweichung von der Norm, bei der Zellen eine unterschiedliche Anzahl von Chromosomen aufweisen, wird Mosaikismus genannt. Mosaik wird bei einigen Frauen mit Turner-Syndrom festgestellt, ist beim Klinefelter-Syndrom jedoch sehr selten .
Äußere Einflüsse.
Nachdem in den 1960er Jahren entdeckt wurde, dass das Medikament Thalidomid schwere Geburtsfehler verursacht, wurde klar, dass viele Medikamente die Plazentaschranke überwinden und den Embryo oder Fötus beeinträchtigen können. In der frühen Embryonalperiode werden die meisten Körperstrukturen gebildet (nach der achten Woche wird der Embryo als Fötus bezeichnet). Obwohl es in der zweiten bis achten Schwangerschaftswoche zu schwerwiegenden körperlichen Defekten kommt, können später bestimmte Anomalien der Augen, des Innenohrs und des Nervensystems auftreten. Bis zur zweiten Woche wird durch die Einwirkung von Schadstoffen die Einnistung des Embryos in die Gebärmutterwand blockiert oder so stark beeinträchtigt, dass die Entwicklung nicht weitergehen kann .
Kinder von Müttern, die während der Schwangerschaft viel getrunken haben, zeigen Anzeichen geistiger und körperlicher Störungen, die als fetales Alkoholsyndrom bekannt sind. Frauen, die während der Schwangerschaft rauchen, haben ein erhöhtes Risiko einer Fehlgeburt, einer Totgeburt oder eines Babys mit niedrigem Geburtsgewicht, bei dem die Wahrscheinlichkeit einer Behinderung oder des Todes deutlich höher ist als bei einem Neugeborenen mit normalem Gewicht.
Auch Spontanaborte, niedriges Geburtsgewicht und andere Probleme stehen im Zusammenhang mit einer schlechten Ernährung der Mutter. Obwohl der Fötus vor vielen Infektionen geschützt ist, können einige davon je nach Entwicklungsstadium, in dem die Infektion aufgetreten ist, schwerwiegende Schäden verursachen. Daher führt die Exposition des Fötus gegenüber dem Rötelnvirus zu Herzfehlern, Blindheit, Taubheit und anderen Störungen (cm. RÖTELN). Einige Infektionen beeinträchtigen den Fötus vor oder während der Geburt und führen zu angeborenen Erkrankungen oder zum Tod. Dazu gehören Zytomegalievirus-Infektionen und Toxoplasmose (beide verlaufen oft mild und für die Mutter unbemerkt) sowie sexuell übertragbare Krankheiten, insbesondere Gonorrhoe, Chlamydien, Herpes genitalis und Syphilis.
Der Embryo oder Fötus kann durch erhöhte ionisierende Strahlung geschädigt werden. Neben der normalen Hintergrundstrahlung ist die Röntgendiagnostik die häufigste Belastungsquelle. Man geht davon aus, dass moderne Diagnosemethoden für Embryo und Fötus ungefährlich sind. Allerdings ist es bei Frauen im gebärfähigen Alter nach Möglichkeit erforderlich, das Becken mit einer Durchleuchtung abzudecken und, sofern nicht unbedingt erforderlich, eine Woche bis zehn Tage nach der Menstruation eine Röntgenaufnahme anzuordnen, da zu diesem Zeitpunkt eine Schwangerschaft unwahrscheinlich ist. Es wurden Bedenken hinsichtlich der Sicherheit nichtionisierender Strahlung von Mikrowellenherden, Computerbildschirmen und diagnostischem Ultraschall geäußert. Bisher konnten diese Bedenken weder aus theoretischer Sicht noch durch statistische Belege bestätigt werden.
Multifaktorielle Ursachen.
Die meisten Geburtsfehler können nicht auf eine genetische Ursache oder einen Umweltfaktor zurückgeführt werden. Es wird angenommen, dass sie entweder das Ergebnis des Zusammenspiels vieler Gene (polygene Ursache) oder der gemeinsamen Wirkung von Genen und Umweltfaktoren (multifaktorielle Ursache) sind.
BEHANDLUNG
Nur sehr wenige angeborene Defekte können vollständig geheilt werden. Durch eine Therapie kann die Entwicklung der meisten von ihnen jedoch verlangsamt oder gestoppt werden, und der resultierende Defekt kann manchmal sogar teilweise korrigiert werden. Strukturelle Defekte wie Lippen-Kiefer-Gaumenspalten (Gaumenspalte), Klumpfuß und verschiedene Defekte des Herzens und des Verdauungstrakts werden operativ korrigiert. Derzeit ist auch die Transplantation verschiedener Organe möglich, darunter Nieren, Leber, Hornhaut und zur Behandlung von Immunschwäche auch Knochenmark. Für defekte oder fehlende Gliedmaßen werden wirksamere Methoden der Prothetik entwickelt. Durch Rehabilitation und sonderpädagogische Methoden können viele geistige und körperliche Auffälligkeiten sowie sensorische Defizite ausgeglichen werden. Einige angeborene Stoffwechselstörungen können durch Diät oder Medikamente behandelt werden.
Kinder mit angeborener Hypothyreose entwickeln sich normal, wenn ihnen rechtzeitig Schilddrüsenhormone verabreicht werden. Eine spezielle Diät kann die meisten Kinder mit einem so schweren Stoffwechseldefekt wie der Phenylketonurie vor verheerenden Hirnschäden bewahren (siehe PHENYLKETONURIE). Bei erblich bedingter Rachitis werden Vitamin-D- und Phosphatpräparate erfolgreich eingesetzt. Erkrankungen, die durch übermäßige Flüssigkeitsansammlung entstehen, insbesondere Hydrozephalus und Harnwegsverstopfungen, können operativ behandelt werden, in manchen Fällen sogar schon vor der Geburt.
Fortschritte wurden bei der Behandlung in der pränatalen Phase und bei der Anwendung nicht-chirurgischer Methoden erzielt. Funktionsstörungen des Herzens werden mit Hilfe von Medikamenten korrigiert, die die Mutter erhält, und bei Stoffwechselstörungen im Zusammenhang mit Vitaminmangel werden der Mutter hohe Dosen des benötigten Vitamins verschrieben. Mittlerweile wurden Impfstoffe entwickelt, um Geburtsfehlern aufgrund von Röteln und Rh-Inkompatibilität vorzubeugen, die auftreten, wenn die Antikörper einer Rh-negativen Mutter zerstört werden RotBlutzellen von ihrem Rh-positiven Fötus (cm. BLUT) .
ERKENNUNG UND DIAGNOSE
Mit biochemischen Methoden werden eine Reihe genetischer Erkrankungen von Neugeborenen identifiziert. Einige davon, darunter Hypothyreose, Phenylketonurie und Galaktosämie (eine Störung des Kohlenhydratstoffwechsels), können durch eine Blutuntersuchung aus der Ferse eines Neugeborenen festgestellt werden. Eine rechtzeitige medikamentöse Behandlung oder eine spezielle Diät sorgt für eine normale Entwicklung kranker Kinder.
Für verheiratete Paare, die den Verdacht haben, dass ihr Kind eine genetische Erkrankung haben könnte, gibt es eine medizinisch-genetische Beratung. In der Regel wünschen Paare eine Beratung, weil sie bereits ein Kind mit einem Geburtsfehler haben oder weil sie aufgrund ihrer Familiengeschichte oder ethnischen Zugehörigkeit dem Risiko ausgesetzt sind, ein Kind mit einer bestimmten Erkrankung zu bekommen. Das größte Risiko ist jedoch mit dem Alter der Mutter verbunden – je älter sie ist, desto wahrscheinlicher ist es, dass das Kind an Chromosomenstörungen wie dem Down-Syndrom leidet. Viele Geburtsfehler können jetzt schon im Mutterleib sicher und genau diagnostiziert werden .
Die Ultraschallbildgebung des Fötus gibt Aufschluss über Entwicklungs- und Strukturdefekte und liefert wichtige Informationen über den Verlauf der Schwangerschaft und die bevorstehende Geburt, einschließlich des Gestationsalters, des Vorhandenseins von mehr als einem Fötus, der Position der Plazenta, einer möglichen Herzinsuffizienz des Fötus und der Position des Fötus in der Gebärmutter. Amniozentese, d.h. Die Punktion der Fruchtblase und die Entnahme einer Fruchtwasserprobe (den Fötus umgebende Flüssigkeit) zur Analyse ermöglichen die Erkennung von Chromosomenanomalien, einigen Missbildungen und ca. 100 Stoffwechselstörungen. Die fetale Endoskopie, bei der ein faseroptisches Endoskop in die Gebärmutter eingeführt wird, ist ein schwierigerer und riskanterer Eingriff. Es ermöglicht die Untersuchung des Fötus sowie die Entnahme von Blut- und Gewebeproben für diagnostische Tests. Dieses Verfahren wird auch bei Bluttransfusionen bei Rh-Inkompatibilität angewendet.
Mehr als 95 % der Frauen, die sich einer pränatalen Untersuchung unterziehen, können sicher sein, dass der Fötus nicht an der vermuteten Krankheit leidet. Durch die Information der Eltern wird die Zahl der Abtreibungen drastisch reduziert. Gleichzeitig ermöglichen Informationen über das Vorliegen bestimmter Störungen beim Fötus den Ärzten zum Zeitpunkt der Geburt, sich auf die erforderlichen Maßnahmen vorzubereiten, um das Leben des Neugeborenen zu retten und die schädlichen Folgen seines Defekts zu verringern Informieren Sie die Eltern über zusätzliche Maßnahmen, die zur Erhaltung der Gesundheit des Kindes erforderlich sind.
|
Häufigkeit einiger angeborener Defekte |
|||
|
Krankheit |
Häufigkeit bei der Geburt |
Vererbungstyp 1 |
|
| Erbliche Krankheiten | |||
| Achondroplastischer Kleinwuchs | 1/10 000 | ||
| Mukoviszidose | 1/2000, USA, weiß | ||
| Galaktosämie | 1/30 000–1/40 000 | ||
| Hämophilie A | 1/2500, Männer | ||
| Familiäre Hypercholesterinämie | 1/500 | ||
| Sichelzellenanämie | 1/625, Afroamerikaner | ||
| Tay-Sachs-Krankheit | 1/3600, Juden (Aschkenasen) | ||
| Neurofibromatose | 1/3000 | ||
| Chromosomenanomalien | |||
| Klinefelter-Syndrom | 1/500, Männer | ||
| Turner-Syndrom | 1/10.000, Frauen | ||
| Down-Syndrom | 1/800 | ||
| Angeborene Fehlbildungen | |||
| "Gaumenspalte" | 1/2000 | ||
| Hasenlippe | 1/1150 | ||
| Klumpfuß 2 | 1/400 | ||
| Angeborene Hüftluxation 2 | 1/400 | ||
| Unterentwicklung der Gliedmaßen | 1/2500 | ||
| Spina bifida 3 | 1/2000 | ||
| Herzfehler | 1/200 | ||
| 1 AD – autosomal-dominant; AR – autosomal-rezessiv; XP – X-chromosomal rezessiv. 2 Ohne Defekte des Nervensystems. 3 Ohne Anenzephalie, d.h. das gesamte oder der größte Teil des Gehirns fehlt. Spina bifida ist eine unvollständige Verschmelzung der Spina bifida. |
|||
Die Keimentwicklung (embryonale Entwicklung) eines Menschen ist die frühe Entwicklungsphase bis zu 8 Wochen. In dieser Zeit entsteht aus einer befruchteten Eizelle ein Körper, der alle Grundmerkmale eines Menschen aufweist. Nach achtwöchiger Entwicklung wird der intrauterine Organismus als Fötus bezeichnet, und die Entwicklungsperiode wird als Fötus bezeichnet.
Die menschliche Embryonalentwicklung ist in mehrere Phasen unterteilt. Betrachten wir die Entwicklungsstadien des menschlichen Embryos. Die erste Periode eines einzelligen Embryos (Zygote, die alle Eigenschaften beider Geschlechtszellen besitzt) beginnt mit der Befruchtung der Eizelle, die zwei Kerne enthält. Jeder Kern enthält die Hälfte des Chromosomensatzes (23 Chromosomen vom Vater und 23 Chromosomen von der Mutter).
Der menschliche Embryo beginnt sich langsam entlang des Eileiters zu bewegen, angetrieben durch die Fimbrien des Eileiters und den Flüssigkeitsfluss darin. Das Hauptziel seiner Bewegung ist die Gebärmutter.
Die erste Teilung eines einzelligen menschlichen Embryos erfolgt 30 Stunden nach der Befruchtung. Dann erfolgt die Teilung eine Teilung pro Tag.
Nach vier Tagen nimmt der Embryo das Aussehen eines Klumpens an, der aus etwa 8–12 Zellen besteht. Darüber hinaus erfolgt die Zellteilung des menschlichen Embryos immer schneller.
Während dieser Zeit bereitet sich die Gebärmutter auf die Aufnahme des Embryos vor. Die Schleimhaut der Gebärmutter verdickt sich und wird locker. Darin beginnen viele zusätzliche Blutgefäße zu erscheinen.
Menschliche embryonale Zellen beginnen, Enzyme abzusondern, die die Gebärmutterschleimhaut zerstören. Spezielle Zotten auf der Oberfläche des Embryos beginnen sich schnell zu vermehren und in das Uterusgewebe hineinzuwachsen. Die Implantation eines menschlichen Embryos dauert 40 Stunden. Es entsteht ein neues Organ, die sogenannte Plazenta oder Babystelle. Die Plazenta ist ein Organ, das den Körper der Mutter mit dem Embryo verbindet und den Embryo auch mit Nährstoffen versorgt.
Am Ende der zweiten Woche beträgt die Länge des menschlichen Embryos 1,5 mm.
Die Entwicklung des menschlichen Embryos erfolgt nach einem klar strukturierten Plan.
In der vierten Entwicklungswoche beginnen die Grundlagen der meisten Organe und Gewebe des zukünftigen Menschen zu erscheinen (Nieren, Darm, Knorpel des Wirbelsäulenskeletts, Knochen, quergestreifte Muskulatur, Schilddrüse, Leber, Haut, Ohren, Augen).
In der fünften Woche beträgt die Länge des menschlichen Embryos etwa 7,5 mm. In diesem Stadium können Sie mithilfe einer Ultraschalluntersuchung die Kontraktionen des Herzens des Embryos erkennen.
Während des Zeitraums von 32 Tagen der Entwicklung beginnen sich im menschlichen Embryo die Rudimente der Arme zu entwickeln, und näher am 40. Tag beginnen sich die Rudimente der Beine zu entwickeln.
Am Ende seiner Entwicklung erreicht der Embryo bereits eine Länge von etwa 3-4 cm (vom Scheitel bis zum Steißbein). Zu diesem Zeitpunkt endet die Bildung aller Hauptorgane des Embryos, er erhält alle Merkmale einer Person, sowohl im äußeren Erscheinungsbild als auch in der inneren Organisation.
Bildungsministerium der Russischen Föderation
Bildungsministerium der Stadtverwaltung Tscheboksary
Städtische Bildungseinrichtung „Kadettenschule“
Zusammenfassung zum Thema:
Entwicklung des menschlichen Embryos
Abgeschlossen von: Kadett 9. Klasse „A“.
Ivanov K.
Geprüft von: Nardina S.A.
Tscheboksary 2004
Wie sieht ein Baby ganz am Anfang seines Lebens aus – im Mutterleib?
Das ist ein Ei, also eine Zelle. Sie besteht, wie alle Zellen des menschlichen Körpers, aus einem Substanztröpfchen – Protoplasma mit einem Kern in der Mitte. Dabei handelt es sich um eine sehr große, mit bloßem Auge fast sichtbare Zelle mit einer Größe von einem Zehntel Millimeter.
Dies geschieht durch die Vereinigung zweier Zellen: einer männlichen Zelle, dem Spermium, und einer weiblichen Zelle, der Eizelle. Das Ei ist eine große, abgerundete Zelle. Was das Sperma betrifft, so ist es 30 bis 40 Mal kleiner – allerdings ohne Berücksichtigung seines langen, schwingenden Schwanzes, dank dem sich das Sperma fortbewegt. Bei Kontakt mit der Eizelle verliert das Spermium seinen Schwanz. Und sein Kern dringt in das Ei ein. Beide Kerne verschmelzen, es kommt zur Befruchtung der Eizelle; Von nun an wird sie zu einem Ei. Jede der Zellen, aus denen die Eizelle besteht, trägt die Merkmale eines Elternteils. Träger dieser Eigenschaften sind kleine, stäbchenförmige Strukturen, die in den Kernen aller Zellen enthalten sind und als Chromosomen bezeichnet werden. Der Zellkern jeder Zelle im menschlichen Körper enthält 46 Chromosomen: 23 vom Vater und 23 von der Mutter. Gleichartige Chromosomen von Vater und Mutter bilden ein Paar. Jeder von uns verfügt in jeder Körperzelle über 23 Chromosomenpaare, die für ihn einzigartig sind und seine individuellen Eigenschaften bestimmen; Deshalb ähneln bestimmte Merkmale unseres Aussehens, unseres Geistes oder unseres Charakters unserem Vater und unserer Mutter, unseren Großeltern oder anderen Verwandten.
Das Geschlecht des Kindes ergibt sich aus einer zufälligen Auswahl der Chromosomen. Achten wir zunächst auf das Aussehen der Chromosomen: Ihre Größe und Form sind unterschiedlich, aber in jeder normalen Zelle gibt es mindestens 44 Chromosomen, von denen jedes ein ähnliches hat. Zu zweit gruppiert bilden sie 22 Paare. Sie werden nach Größe klassifiziert: Das größte ist die Nummer 1 und das kleinste ist die Nummer 22. 23 – das Paar unterscheidet sich. Bei einer Frau besteht es wie alle anderen aus zwei ähnlichen Chromosomen, die mit dem Buchstaben X (X) gekennzeichnet sind. Und bei Männern gibt es im 23. Paar nur ein X-Chromosom sowie ein kleineres, das mit dem Buchstaben Y (Y) bezeichnet wird.
Im Körper der Eltern handelt es sich bei der Eizelle oder dem Sperma um Zellen, die nur die Hälfte der Chromosomen enthalten, also jeweils 23. Somit sind alle Eizellen vom gleichen Typ: Sie haben alle ein X-Chromosom. Es gibt zwei Arten von Spermatozoen: Einige von ihnen haben das X-Chromosom Nummer 23, andere haben das Y-Chromosom. Wenn sich eine Eizelle zufällig mit einem Spermatozoen mit einem Somit erfolgt die Geschlechtsbestimmung während der Befruchtung.
Theoretisch wäre es von diesem Moment an möglich, das Geschlecht des Kindes herauszufinden, wenn wir über technische Mittel verfügten, die es uns ermöglichen würden, die Eizelle zu beobachten, ohne das Risiko einer Beschädigung einzugehen. Vielleicht wird der Tag kommen, an dem der Zufall der Wissenschaft weichen wird und die Eltern das Geschlecht ihres Kindes selbst bestimmen werden, auf jeden Fall wird dies nur geschehen, wenn die X- und Y-Spermien im Sperma getrennt werden. Sobald das Ei gebildet ist, beginnt es sich in zwei, vier, acht, sechzehn usw. zu teilen. Zellen. Nach einigen Tagen spezialisieren sich die Zellen funktionell: einige – um Sinnesorgane zu bilden, andere – Eingeweide, andere – Geschlechtsorgane usw. Es ist das Y-Chromosom, das den Keimzellen mitteilt, dass sie sich entsprechend dem männlichen Typ entwickeln werden. Äußere Geschlechtsmerkmale machen sich bereits zu Beginn des vierten Schwangerschaftsmonats bemerkbar. Aber auf der chromosomalen Ebene, die auch seine äußeren Erscheinungsformen bestimmt, existiert Sex vom Moment der Befruchtung an. Deshalb ist es in manchen Fällen möglich, das Geschlecht des Kindes bereits zu Beginn der Schwangerschaft (im zweiten oder dritten Monat) durch Chromosomenuntersuchungen einiger Eizellen (sogenannte Trophoblastenpunktion) herauszufinden Amniozentese) oder dank einer Art Radar, das es Ihnen mithilfe von Ultraschall ermöglicht, einen kleinen Penisembryo in der Gebärmutter der Mutter zu sehen.
Die befruchtete Eizelle bewegt sich durch den Eileiter, teilt sich gleichzeitig und verwandelt sich in einen mehrzelligen Embryo, der nach 4-5 Tagen in die Gebärmutterhöhle gelangt. Der Embryo bleibt 2 Tage lang frei in der Gebärmutter, sinkt dann in die Schleimhaut ein und heftet sich daran fest. Die embryonale Periode der intrauterinen Entwicklung beginnt. Einige Zellen bilden Membranen. Die äußere Hülle hat Zotten mit Kapillaren. Die Ernährung und Atmung des Embryos erfolgt über die Zotten. Im Inneren der Zottenhülle befindet sich eine weitere, dünn und durchsichtig, wie Zellophan. Es bildet sich eine Blase. Ein Embryo schwimmt in der Flüssigkeit der Blase. Diese Hülle schützt den Embryo vor Stößen und Lärm.
Am Ende des 2. Monats der intrauterinen Entwicklung verbleiben die Zotten nur noch auf der der Gebärmutter zugewandten Seite der Embryonalmembran. Diese Zotten wachsen und verzweigen sich und dringen in die Gebärmutterschleimhaut ein, die reichlich mit Blutgefäßen versorgt ist. Die Plazenta entwickelt sich in Form einer Scheibe und ist fest in der Gebärmutterschleimhaut verankert. Von diesem Moment an beginnt die fetale Phase der intrauterinen Entwicklung.
Über die Wand der Blutkapillaren und Plazentazotten werden Gase und Nährstoffe zwischen Mutter und Kind ausgetauscht. Das Blut von Mutter und Fötus vermischt sich nie. Ab dem 4. Schwangerschaftsmonat schüttet die Plazenta als endokrine Drüse ein Hormon aus. Dadurch löst sich die Gebärmutterschleimhaut während der Schwangerschaft nicht ab, es treten keine Menstruationszyklen auf und der Fötus bleibt während der gesamten Schwangerschaft in der Gebärmutter.
Wenn zwei oder mehr Eier ovuliert werden, werden zwei oder mehr Föten gebildet. Das sind zukünftige Zwillinge. Sie sind einander nicht sehr ähnlich. Manchmal entwickeln sich zwei Föten aus derselben Eizelle, und oft teilen sie sich dieselbe Plazenta. Solche Zwillinge haben immer das gleiche Geschlecht und sind einander sehr ähnlich
Der Embryo entwickelt sich schnell in der Gebärmutter. Am Ende des ersten Monats der intrauterinen Entwicklung beträgt der Kopf des Fötus 1/3 der Körperlänge, die Konturen der Augen erscheinen und in der 7. Woche können Finger unterschieden werden. Nach 2 Monaten wird der Embryo menschenähnlich, obwohl seine Länge zu diesem Zeitpunkt 3 cm beträgt.
Nach 3 Monaten intrauteriner Entwicklung sind fast alle Organe gebildet. Zu diesem Zeitpunkt kann das Geschlecht des ungeborenen Kindes bestimmt werden. Mit 4,5 Monaten sind Kontraktionen des fetalen Herzens zu hören, deren Häufigkeit doppelt so hoch ist wie die der Mutter. Während dieser Zeit wächst der Fötus schnell und wiegt im Alter von 5 Monaten etwa 500 g und zum Zeitpunkt der Geburt 3 bis 3,5 kg.
REFERENZLISTE:
1. Enzyklopädie Blinov I.I. und Karzova S.V. S. 367–369
2.Lehrbuch über Biologie für die 9. Klasse, Autor: Tsuzmer A.M., Petrishina O.L. S. 167–172
Zusammensetzung des menschlichen Embryos in den ersten Tagen seiner Existenz
Befruchtung der Eizelle – Seite 3
Bildung der Plazenta - Seite 3
Entwicklung des Embryos – Seite 4
Referenzen – Seite 5

Die Wirkstoffe gelangen über den extravaskulären Weg von der Plazenta zum Embryo und erfüllen somit eine Schutzfunktion. Basierend auf dem Vorstehenden können wir die Hauptmerkmale der frühen Entwicklungsstadien des menschlichen Embryos feststellen: 1) asynchroner Typ der vollständigen Fragmentierung und Bildung von „hellen“ und „dunklen“ Blastomeren; 2) frühe Trennung und Bildung extraembryonaler Organe; 3) frühe Bildung der Fruchtblase und...





Die Embryonalperiode ist ein zweischichtiger Schild, der aus zwei Schichten besteht: der äußeren Keimschicht (Ektoderm) und der inneren Keimschicht (Endoderm). Abb.2. Die Position des Embryos und der Keimhäute in verschiedenen Stadien der menschlichen Entwicklung: A - 2-3 Wochen; B – 4 Wochen: 1 – Amnionhöhle; 2 - Körper des Embryos; 3 - Dottersack; 4 - Tropholast; B - 6 Wochen; G - Fötus 4-5 Monate: 1 - Embryokörper...
Das Problem wurde von Engels in seinem 1896 veröffentlichten Werk „Die Rolle der Arbeit im Prozess der Transformation vom Affen zum Menschen“ offengelegt, obwohl es kurz nach der Veröffentlichung von Darwins „Die Abstammung des Menschen“ geschrieben wurde. Zu dieser Zeit verfügte die Wissenschaft über relativ wenige Daten über die fossilen Vorfahren des Menschen. Später bestätigten zahlreiche Funde von Überresten von Knochen und Werkzeugen fossiler Menschen auf brillante Weise ...

Fünf Gehirnbläschen (Hohlräume mit Flüssigkeit); Es gibt auch hervortretende Augen mit Linsen und pigmentierter Netzhaut. Im Zeitraum von der fünften bis achten Woche endet die eigentliche Embryonalperiode der intrauterinen Entwicklung. In dieser Zeit wächst der Embryo von 5 mm auf etwa 30 mm und beginnt, einem Menschen zu ähneln. Sein Aussehen ändert sich wie folgt: 1) Die Biegung nimmt ab...